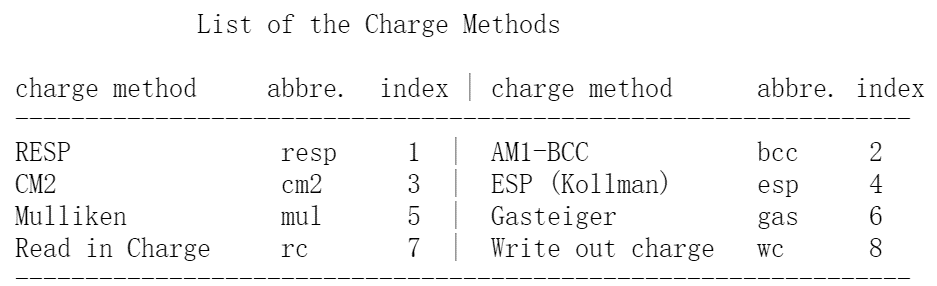
antechamber -i SUB.pdb -fi pdb -o SUB.mol2 -fo mol2 -c bcc -pf y
输出.mol2文件
-c bcc: use the AM1-BCC charge model in order to calculate the atomic point charges.
parmchk -i SUB.mol2 -f mol2 -o SUB.frcmod
输出成.frcmod
.frcmod: a parameter file that can be loaded into LEaP in order to add missing parameters.
you may see any parameters listed with the comment “ATTN: NEEDS REVISION” then it means that Antechamber could not determine suitable parameters and so you must manually provide these before you can proceed with the simulation.
source leaprc.ff99SB source leaprc.gaff SUB = loadmol2 SUB.mol2 check SUB loadamberparams SUB.frcmod saveoff SUB SUB.lib saveamberparm SUB SUB.prmtop SUB.inpcrd quit
Creating topology and coordinate files for enzyme_substrate complex
加H,并对HIS, ASP, GLU和二硫键SSBOND进行判断和修改
a. 加氢的软件有很多,antchamber里面的加氢程序一般,而且产生的输出文件有很多需要手动调整的地方。可以选用其他软件如:薛定谔,PDB2PQR Server等。此处我用的是后者PDB2PQR Server (https://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/).
b. HIS有3种质子化状态(HID,HIE,HIP), ASP质子化为ASH, GLU质子化为GLH.(详见word文档 “Amber蛋白质力场中组氨酸相关参数的识别与选择”).
c. 有的时候后面进行的程序会报错还需要注意修改末端氨基酸报错原子的命名方式,必要时在ATOM和HETATM之间空一行添加词"TER".
d. SSBOND一定要在开头和结尾加上SSBOND连接的原子标号和CONNECT连接关系:
1 2 3 4 5 6 7 8 9
SSBOND 1 CYX A 25 CYX A 264 SSBOND 2 CYX A 36 CYX A 39 SSBOND 3 CYX A 231 CYX A 240 … TER END CONECT 538 584 CONECT 374 4018 CONECT 3545 3663
$tleap -f leaprc.ff99SB
加载tleap (不需要复制$)
source leaprc.gaff
加载小分子力场gaff
ensure the GAFF force field is available
loadoff SUB.lib
加载第1步"Creating topology and coordinate files for ligand"里建立的小分子SUB库
solvatebox complex TIP3PBOX 12
solvatebox solute solvent buffer [ “iso” ] [ closeness ]
In this case our solute is the NMA unit we created. For the solvent we will use TIP3PBOX. This is a pre-equilibrated box of TIP3P water. Type “edit TIP3PBOX” if you want to take a look at it.
For buffer we will specify 15 which means we want a solvent box of 15 angstroms between any atom in our NMA and the edge of the box.
跑MD之前,我们需要对conf文件按需修改一下,其中有两部分参数(#periodic boundry conditions#和#Fix residues and sub in the QM region#)的修改需要计算一下。
periodic boundry conditions中x(cellbasisvector1), y(cellbasisvector2), z(cellbasisvector3)以及cellorigin的计算方法
VMD加载第3步"Creating waterbox for enzyme_substrate complex"做好水盒子的AMBER.pdb后,在TKconsole依次输入下面每一行代码:
1 2 3 4 5
set boxsize [open boxsize.dat w] set everyone [atomselect top all] puts $boxsize "[measure minmax $everyone]" puts $boxsize "[measure center $everyone]" close $boxsize
Fix residues and sub in the QM region
上一步做完后,接着输入一下代码($不能省略):
1 2 3 4 5
set everyone [atomselect top all] $everyone set beta 0 set prolig [atomselect top "resid 78 140 141 199 253 266"] $prolig set beta 1 $everyone writepdb prolig-fix.pdb
该步的意思就是将AMBER.pdb中78, 140, 141, 199, 253氨基酸和266底物的最后一列的值由0变为1供namd识别为fix residues and sub in the QM region,并保存为prolig-fix.pdb.
(p.s. "Calculating the parameter of waterbox"中两步操作的代码可以通过合并建立一个namd-prep.tcl脚本在TKconsole里面执行vmd -dispdev text -e namd-prep.tcl,但是我个人的电脑或者在超算上这么执行就会卡住,所以一般我都是手动输入代码执行。)
1 2 3 4 5 6 7 8 9 10 11 12 13
##measuere the size of system## set boxsize [open boxsize.dat w] set everyone [atomselect top all] puts $boxsize "[measure minmax $everyone]" puts $boxsize "[measure center $everyone]" close $boxsize
##fix protein, ion (PDB) and ligand## set everyone [atomselect top all] $everyone set beta 0 set prolig [atomselect top "resid 78 140 141 199 253 266"] $prolig set beta 1 $everyone writepdb prolig-fix.pdb
############################################################# ## Simulation parameters ## ############################################################# #insert files and force field(section 3.2.1)########### amber on #specifies we are using AMBER Prm and CRD files parmfile AMBER.prmtop #specifies we are using AMBER Prm and CRD files ambercoor AMBER.inpcrd #specifies we are using AMBER Prm and CRD files
#output file (section 3.2.2)############# outputname $outputname #specifies the prefix for the output files binaryoutput no #activates binary output (yes=binary) DCDfile $outputname.dcd #specifies the output file name for coordinates trajectory DCDfreq 5000 #number of timesteps between the writing coordinates restartname $outputname.restart #specifies the restart filename restartfreq 5000 #number of timesteps between the writing coordinates
#standard output (section 3.2.3)################ outputEnergies 5000 #number of timesteps between energy output outputTiming 5000 #number of timesteps between timing output outputMomenta 5000 #number of timesteps between momenta output outputPressure 5000 #number of timesteps between pressure output
#timestep parameters (section 7.3.1)########## firsttimestep 0 #frame number where the simulation starts timestep 2 #length of each step in fs stepspercycle 10 #how often the interacting particle lists are updated
#space partitionin (section 5.3.2)################# cutoff 12 #non-bonded cutoff switching off #switch off for Amber #switchdist 10 #distance at which the smoothing function is applied pairlistdist 14 #distance >= switchdist for atom pairs to be included in the pairlist margin 0 #extra length in patch dimensions
#basic dynamics (section 5.3.3)################ exclude scaled1-4 #which bonded atom pairs are excluded from non-bonded calculations temperature $temperature #INITIAL temperature of the simulation note delete on restart 1-4scaling 0.833333 #1.0 for Charmm, 0.833333 for Amber scnb 2 #This is default for both Amber and Charmm rigidbonds all #controls how shake is used rigidTolerance 0.00001 #allowable bond length error for shake nonbondedFreq 1 #number of timesteps between nonbond evaluation fullElectFrequency 2 #number of timesteps between full electrostatic evaluations
#PME parameters (section 5.3.5)############### PME yes #turns PME on or off (yes=on no=off) PMEGridSpacing 1.0 #standard spacing. Namd asigns the grid size.
#periodic boundry conditions (section 6.4.3)############# cellbasisvector1 75.184 0 0 #defines the first periodic boundary cellbasisvector2 0 73.832 0 #defines the second periodic boundary cellbasisvector3 0 0 77.557 #defines the third periodic boundary cellorigin -0.066 0.047 -0.147 #defines the xyz location of the center of the box
#defines file which contains the PBC info from previous runs########### wrapwater on #wraps the waters to one box. wrapall on #wraps every contiguous cluster of bonded atoms wrapNearest on #wraps coordinates to the nearest image to the origin
####Fix residues and sub in the QM region########## fixedAtoms on fixedAtomsForces on fixedAtomsFile prolig-fix.pdb fixedAtomsCol B
#temperature control and equilibration (section 6.3)########## langevin on #turns langevin dynamics on or off langevinTemp $temperature #temp to which langevin dynamics will be adjusted langevinDamping 5 #damping coefficient for langevin dynamics langevinHydrogen off # don't couple langevin bath to hydrogens
###pressure control(section 6.3)########## langevinPiston on #turns on constant pressure langevinPistonTarget 1.0325 #target pressure langevinPistonPeriod 100 #period over which the pressure is averaged in fs(oscillation time constant) langevinPistonDecay 50 #damping time scale in fs langevinPistonTemp $temperature #should be equal to langevin temp ###minimization########### minimize 5000